Pan-phylum genomes of hornworts revealed conserved autosomes but dynamic accessory and sex chromosomes
Last Updated: 2024-11-27
Table of Contents
Abstract
Hornworts, one of the three bryophyte phyla, represent some of the deepest divergences in extant land plants, with some families separated by more than 300 million years. Previous hornwort genomes represented only one genus, limiting the ability to infer evolution within hornworts and their early land plant ancestors. Here we report ten new chromosome-scale genomes representing all hornwort families and most of the genera. We found that despite the deep divergence, synteny was surprisingly conserved across all hornwort genomes, a pattern that might be related to the absence of whole genome duplication. We further uncovered multiple accessory and putative sex chromosomes that are highly repetitive and CpG methylated. In contrast to autosomes, these chromosomes mostly lack syntenic relationship with one another and are evolutionarily labile. Notable gene retention and losses were identified, including those responsible for flavonoid biosynthesis, stomata patterning, and phytohormone reception, which have implications in reconstructing the evolution of early land plants. Together, our pan-phylum genomes revealed an array of conserved and divergent genomic features in hornworts, highlighting the uniqueness of this deeply diverged lineage.
The following are detailed methods used in analysis of hornwort genomes.
Dependencies and Scripts
| Name | Purpose | Citation |
|---|---|---|
| addFastaMetadata.py | Add metadata fields to fasta sequence ids | Custom |
| auN.py | Calculate and graph “area under” curves for assessing genome contiguiuty | Custom |
| convertFastqToFasta.py | SeqIO wrapper to convert files | Custom |
| extract_UnknownLTR.py | Get LTR sequences classified as unknown type from EDTA TE library | Custom |
| getFastaSeqLengths.py | Custom | |
| getFromFasta.py | Extract specific sequence(s) from a fasta file | Custom |
| pilonIterative.sh | Iteratively run pilon to polish genome a given number of times | Custom |
| pyTable.py | Count amounts of each unique item in a single-column file | Custom |
| removeAlternativeTranscripts.py | Produce primary transcript file containing only the longest transcript for each gene | Custom |
| renameFasta.py | Convert sequence ids in a fasta file using a conversion table | Custom |
| renameFastaAndReorder.py | Convert sequence ids in a fasta file and reorder them using a conversion table | Custom |
| renameGTF_Phytozome.py | Convert gene and transcript ids in a GTF file using a conversion table | Custom |
| summaryStats.R | Generate summary stats for a list of numbers piped to script through STDIN | Custom |
| trf2gff.py | Convert tandemrepeatsfinder output to GFF format | Custom |
| flye 2.8 | Long-read assembly | Kolmogorov et al. 2019 |
| Pilon 1.24 | Draft assembly polishing | Walker et al. 2014 |
| TGS-Gapcloser 1.1.1 | Filling gaps in scaffolded assembly | Xu et al. 2020 |
| juicer | Hi-C analysis | Durand et al. 2016 |
| Juicebox Assembly Tools | Hi-C visualization | Aiden Lab |
| hicexplorer 3.7.2 | Hi-C visualization and analysis | Ramirez et al. 2018 |
| pyGenomeTracks 3.8 | Genome visualization | Lopez-Delisle et al. 2021 |
| bwa 0.7.17-r1188 | Short-read mapping | Li 2013 |
| minimap2 2.17-r941 | Long-read mapping | Li 2018 |
| hisat2 2.2.1 | RNA read mapping | Kim et al. 2019 |
| fastp 0.20.0 | Short-read adapter/low quality base trimming | Chen et al. 2018 |
| porechop 0.2.4 | ONT read adapter trimming | Wick 2018 |
| EDTA 2.0.1 | Repeat annotation and analysis | Ou et al. 2019 |
| Tandem Repeats Finder 4.09.1 | Tandem repeat identification | Benson 1999 |
| BRAKER 2.1.5 | Gene prediction | Bruna et al. 2021 |
| EggNOG mapper 2.1.9 | Gene function annotation | Cantalapiedra et al. 2021 |
| OrthoFinder 2.5.4 | Gene orthogroup inference | Emms and Kelly 2019 |
| GENESPACE 1.3 | Synteny analysis | Lovell et al. 2022 |
| wgd 1.0 | Whole genome duplication analysis | Zwaenepoel and Van de Peer 2019 |
| CAFE 5 | Orthogroup expansion/contraction | Mendes et al. 2020 |
| r8s | Divergence time estimation | Sanderson 2003 |
| gffread 0.11.7 | GFF file manipulation | Pertea and Pertea 2020 |
| AGAT | GTF/GFF file conversion | Dainat 2023 |
| IQ-TREE 2.0.3 | Phylogenetic inference | Minh et al. 2020 |
| BUSCO 5.2.1 | Assembly completeness test | Manni et al. 2021 |
| BlobToolKit 4.1.2/Blobtools2 | Assembly contamination identification | Challis et al. 2020 |
| BLAST+ toolkit 2.10.0 | Sequence similarity search | Camacho et al. 2009 |
| DIAMOND 2.0.15 | Sequence similarity search | Buchfink et al. 2021 |
| Megalodon 2.5.0 | ONT modified methylation calling | Github |
| bismark 0.24.1 | bisulfite-seq methylation calling | Krueger et al. 2011 |
| RepeatMasker 4.1.0 | Masking repetitive genomic sequence | Smit et al. 2013-2015 |
| Stringtie 2.1.1 | Transcriptome assembly | Kovaka et al. 2019 |
| Ballgown | Gene differential expression | Frazee et al. 2015 |
| goatools | GO Term enrichment | Klopfenstein et al. 2018 |
| pfam_scan | Protein-family domain annotation | EMBL |
Methods
SequencingTop
High molecular weight DNA was sequenced on Oxford Nanopore R9 MinION flowcells and basecalled with Guppy v5 using the dna_r9.4.1_450bps_sup model.
AssemblyTop
ONT reads less than 5 kbp were removed and the remainder were assembled with Flye v2.9:
awk 'BEGIN {FS = "\t" ; OFS = "\n"} {header = $0 ; getline seq ; getline qheader ; getline qseq ; if (length(seq) >= 5000) {print header, seq, qheader, qseq}}' < your.fastq > filtered.fastq
flye --nano-hq filtered.fastq -t 24 -o flye
Contigs were corrected with Illumina DNA sequence data using Pilon v1.24 in three iterations, with the Pilon output as input each successive round:
bwa index assembly.fasta
bwa mem -t 24 assembly.fasta Illumina_reads_R1.fq Illumina_reads_R2.fq | samtools sort -o illumina.bam
minimap2 -t 24 assembly.fasta ONT_reads.fq | samtools sort -o ont.bam
java -Xmx50G -jar pilon-1.24.jar --genome assembly.fasta --frags illumina.bam --nanopore ont.bam --output pilon
Three rounds of Pilon generally made >95% of all potential changes with diminishing returns (and possible over-polishing errors) with further rounds.
ScaffoldingTop
HiC libraries were prepared, sequenced, and scaffolded by Phase Genomics (Seattle, WA). TGS-Gapcloser was used to fill gaps between scaffolds with ONT reads and polish filled gaps with Illumina data:
convertFastqToFasta.py ONT_reads.fq
cat Illumina_reads_R1.fq Illumina_reads_R2.fq > Illumina_reads_combined.fq
tgsgapcloser --scaff scaffolded_assembly.fasta \
--reads ONT_reads.fasta \
--ouput assembly.gapclosed
--ngs Illumina_reads_combined.fq \
--pilon /home/ps997/bin/pilon-1.24.jar \ # Change to match your system
--samtools /usr/local/bin/samtools \ # Change to match your system
--java /usr/bin/java # Change to match your system
DecontaminationTop
Scaffolded assemblies were checked for contamination using a combination of HiC contact heatmaps and BlobTools2. Illumina WGS reads were mapped to each genome with bwa; scaffold sequences were BLASTed against the NCBI nt database. BAM alignment and BLAST output were added into a BlobDir along with each genome.
bwa mem -t 24 scaffolded_assembly.fasta reads_1.fastq.gz reads_2.fastq.gz | samtools sort -o scaffolded_assembly.readsMapped.bam
blastn -db nt \ # Database needs to be downloaded from NCBI. Change path to match your system
-query scaffolded_assembly.fasta \
-outfmt "6 qseqid staxids bitscore std" \
-max_target_seqs 10 \
-max_hsps 1 \
-evalue 1e-25 \
-num_threads 16 \
-out blast.out
blobtools create \
--fasta scaffolded_assembly.fasta \
--cov assembly.reads.bam \
--hits blast.out \
--taxrule bestsumorder \
--taxdump ~/taxdump \ # File needs to be downloaded from NCBI ftp://ftp.ncbi.nih.gov/pub/taxonomy/new_taxdump/new_taxdump.tar.gz and unpacked. Change path to match your system
AssemblyName
blobtools view --interactive
Open an interactive viewer. The coverage vs. %GC vs. BLAST hit graph is most informative – look for obvious outliers. I require at least two of the three criteria to differ from the primary genome to call a likely contaminant sequence. Then check likely contaminants in the HiC heatmap with JuiceBox Assembly Tools (JBAT). Contaminants should show no contact with the primary genome. If there is contact, consider other explanations e.g. HGT, misassembly. A new FASTA get be made using a custom script:
getFromFasta.py scaffolded_assembly.fasta scaffold100 > contaminant.fasta
getFromFasta.py -v scaffolded_assembly.fasta scaffold100 > scaffolded_assembly.no_contaminants.fasta
Repeat AnnotationTop
Repeats were identified in a first pass by EDTA v2:
EDTA.pl --sensitive 1 --anno 1 --evaluate 1 -t 12 \
--genome scaffolded_assembly.fasta \
--repeatmasker /home/ps997/bin/RepeatMasker/RepeatMasker \ # Change to match your system
--cds ~/HornwortBase_20210503/SPECIES_CDS.fna \ # OPTIONAL: I used these because I already had predicted CDS sequences from a previous round of annotation
&> edta.out # Capture the output for potential debugging
NOTE: EDTA doesn’t like long sequence names, you might have to rename them first with some simple Python code:
infile = open("scaffolded_assembly.fasta","r")
outfile = open("scaffolded_assembly.renamed.fasta","w")
counter = 1
for line in infile:
if line.startswith(">"):
outfile.write(">%s\n" % counter)
counter += 1
else:
outfile.write("%s" % line)
OPTIONAL: EDTA is often used as a stand-alone tool these days, but some additional processing may improve the transposable element (TE) library. These steps aim to recover protein-coding genes that were misidentified as LTRs, following steps from the MAKER wiki. Search databases are linked at the bottom of that page.
# Pull out LTRs not identified as Copia or Gypsy type from the EDTA TE library
extract_unknownLTR.py scaffolded_assembly.renamed.fasta.mod.EDTA.TElib.fa
# BLAST search the unknown LTRs against a curated database of transposons
blastx -query scaffolded_assembly.renamed.fasta.mod.EDTA.TElib.fa.LTRlib.unknown.fa -db Tpases020812 -evalue 1e-10 -num_descriptions 10 -out scaffolded_assembly.renamed.fasta.mod.EDTA.TElib.fa.LTRlib.unknown.fa.blastx -num_threads 12
# Parse the BLAST results and identify unknown LTRs with hits to known transposons
perl Custom-Repeat-Library/transposon_blast_parse.pl --blastx scaffolded_assembly.renamed.fasta.mod.EDTA.TElib.fa.LTRlib.unknown.fa.blastx --modelerunknown scaffolded_assembly.renamed.fasta.mod.EDTA.TElib.fa.LTRlib.unknown.fa
# Combine the LTRs that have BLAST hits to the main LTR library file. Rename the unknown LTRs file.
cat scaffolded_assembly.renamed.fasta.mod.EDTA.TElib.fa.LTRlib.known.fa identified_elements.txt > scaffolded_assembly.renamed.fasta.mod.EDTA.TElib.fa.LTRlib.known.final.fa
mv unknown_elements.txt scaffolded_assembly.renamed.fasta.mod.EDTA.TElib.fa.LTRlib.unknown.final.fa
# BLAST search the LTR library against UNIPROT plant protein database.
blastx -db uniprot_sprot_plants.fasta -query scaffolded_assembly.renamed.fasta.mod.EDTA.TElib.fa.LTRlib.known.final.fa -out uniprot_plant_blast.out -num_threads 12
# Parse the BLAST results and remove LTRs that matched plant proteins.
perl ProtExcluder1.1/ProtExcluder.pl uniprot_plant_blast.out scaffolded_assembly.renamed.fasta.mod.EDTA.TElib.fa.LTRlib.known.final.fa
# Mask the repeats in the genome using this new LTR library. The `--xsmall` option softmasks the genome, which is preferred by BRAKER.
/home/ps997/bin/RepeatMasker/RepeatMasker -noisy -a -gff -u -pa 24 --xsmall -lib scaffolded_assembly.renamed.fasta.mod.EDTA.TElib.fa.LTRlib.known.final.fanoProtFinal scaffolded_assembly.renamed.fasta
# Calculate stats
RepeatMasker/util/buildSummary.pl -useAbsoluteGenomeSize scaffolded_assembly.renamed.fasta.out > scaffolded_assembly.renamed.fasta.repeat-summary.txt
Tandem Repeats Finder
The software tandem repeats finder (TRF) can find additional tandem repeats that are associated with centromeres and telomeres. To avoid double-counting repeats already identified by EDTA/RepeatMasker, we use a hard-masked version of the genome output by RepeatMasker.
Run TRF:
trf scaffolded_assembly.renamed.fasta.masked 2 7 7 80 10 50 2000 -h -d -m -ngs > trf_out.txt
Convert TRF output to GFF, then combine with the repeats GFF file produced by EDTA/RepeatMasker and sort so they are in genomic order.
trf2gff.py trf_out.txt 50 > trf_out_min50.gff
awk -F"\t" '{print $1"\t"$2"\ttandem_repeat\t"$4"\t"$5"\t"$6"\t"$7"\t"$8"\t"$9}' trf_out_min50.gff > trf_out_min50.renamed.gff
cat other_repeats.gff trf_out_min50.renamed.gff | sort -k1,1 -k 4,4n > all_repeats.gff
Gene PredictionTop
Gene models were predicted using a developement version of BRAKER3 (), with input consisting of Illumina RNA reads mapped to the softmasked genome using HISAT2 () and predicted hornwort proteins from published Anthoceros genomes (Li et al 2020, Zhang et al. 2020). BRAKER output files were screened for genes with in-frame stop codons, which were marked as pseudogenes in the corresponding GTF file. Genes were renamed to contain their respective scaffold/contig name plus a number incremented by 100, restarting at the beginning of each scaffold/contig. Subsets of primary transcripts were created by selecting the longest transcript associated with each gene.
Code for mapping RNA reads and running BRAKER and processing output
hisat2-build -p 12 scaffolded_assembly.renamed.fasta.masked.fasta scaffolded_assembly.renamed.fasta.masked.fasta
hisat2 -p 12 -x scaffolded_assembly.renamed.fasta.masked.fasta -1 RNA_R1.fq.gz -2 RNA_R2.fq.gz 2> hisat-align.out | samtools sort -o scaffolded_assembly.renamed.fasta.masked.RNAmapped.bam
braker.pl \
--genome scaffolded_assembly.renamed.fasta.masked.fasta \
--bam scaffolded_assembly.renamed.fasta.masked.RNAmapped.bam \
--prot_seq Hornwort_orthogroups.faa \ # Protein sequences from shared orthogroups from previously published hornwort genomes
--prg=gth \
--gth2traingenes \
--verbosity 3 \
--cores 12 \
--nocleanup \
--softmasking
Filter genes with in-frame stop codons
BRAKER will sometimes predict proteins that contain in-frame (internal) stop codons. In the BRAKER-produced CDS/AA FASTA files, the sequences in the bad regions are masked with N (CDS) or X (AA), but the GTF file will still contain annotation that creates a bad sequence. Following NCBI protocol for genes that are ‘broken’ but are not thought to be pseudogenes, these will get annotated with pseudo=true. The easiest starting point is the bad_genes.lst if you ran BRAKER with the --nocleanup option. If you didn’t, make a new translation from the GTF:
gffread -y proteins.fasta -g genome.fasta augustus.hints.gtf
Then search for sequences with periods (representing stop codons), and write them to bad_genes.lst
with open("proteins.fasta", "r") as infile, open("bad_genes.lst", "w") as outfile:
bad_genes = []
for line in infile:
if line.startswith(">"):
seqid = line.strip(">|\n")
elif "." in line and seqid not in bad_genes:
bad_genes.append(seqid)
outfile.write("%s\n" % seqid)
Rename Contigs and Genes
You’ll probably want to rename the contigs and genes in the fasta and gff/gtf files associated with each genome. The input genome and any annotations must match (e.g. can’t use RepeatModeler annotations if you renamed the genome for EDTA). Here it may be advisable to add a unique ID or version number to link this particular genome assembly and annotation. Comment lines can be added to the header of the final GFF/GTF and fasta metadata can be added to sequence names. In this example, I start with the genome that was temporarily renamed to run EDTA.
Naming conventions for hornwort genomes:
- Scaffolds: Two letters of genus + three letters of species (optional information after this e.g cultivar, sex) + period + S/C (for scaffold or contig) + number (order determined by longest to shortest length).
- Genes: Scaffold ID + G + six digit number unique to each gene. Genes increment by 100 and numbering restarts for each sequence.
- Transcripts: Gene ID + "." + transcript number (increments by 1).
# Get the length of all sequences in order to rename from shortest to longest
getFastaSeqLengths.py PGA_assembly.gapcloser.scaff_seqs.renamed.fasta
sort -k2,2nr PGA_assembly.gapcloser.scaff_seqs.renamed.fasta_sequence_lengths.tmp > genome.fa_sequence_lengths.tsv.sorted.tmp
# Change "j" to the prefix of the names that will be the same in all sequences. Change "id" if using a unique ID for this genome and its annotations
awk -F"\t" -v i="1" -v j="AnagrOXF.C" -v id="v1" '{ print $1"\t"j""i++" id="id }' genome.fa_sequence_lengths.tsv.sorted.tmp > genome.fa_sequence_lengths.tsv.new_contig_names.tsv
### NOTE: Here I manually edit the *new_contig_names.tsv file to change scaffold/contig designations
renameFastaAndReorder.py PGA_assembly.gapcloser.scaff_seqs.renamed.fasta genome.fa_sequence_lengths.tsv.new_contig_names.tsv
# Rename a BRAKER GTF file
renameGTF_Phytozome.py -i augustus.hints.gtf --contig-table genome.fa_sequence_lengths.tsv.new_contig_names.tsv --assembly-id 6cde96438c2e713efa5c285e4fe3a62d
Create pseudo-gene annotated GTF
Run renameGTF_Phytozome.py with --bad-genes bad_genes.lst. It will add pseudo=true to the GTF file to mark broken genes in NCBI style.
Create primary transcript file
This creates a new file with the longest transcript for each gene. Only works assuming sequence IDs end in “.tn” and everything preceeding is a unique gene ID. Depending on the gene prediction software, “.t1” transcripts may be decided as the transcript with the highest support. However, we found that using only “.t1” transcripts as the primary transcripts created a decrease in complete BUSCOs of several percentage points, whereas using the longest transcripts did not have this effect.
removeAlternativeTranscripts.py braker.faa
Functional AnnotationTop
Gene functions were predicted using the eggNOG mapper tool by comparison to the eggNOG 5.0 database using default search settings.
emapper.py -i braker.faa -o species_name
Domains within proteins were also annotated by pfam_scan, using the amino acid sequences as input.
pfam_scan.pl -fasta braker.faa \
-dir ~/pfamDB/ \ # Change to wherever you downloaded your PFAM databse
-cpu 24 \
> braker.faa.pfams
MethylationTop
Methylation was called using Oxford Nanopore Technology’s megalodon:
megalodon ../nanopore_reads/ --guppy-config dna_r9.4.1_450bps_hac.cfg --remora-modified-bases dna_r9.4.1_e8 hac 0.0.0 5mc CG 0 --outputs basecalls mappings mods --reference ../genome/genome.fasta --devices 0 --processes 12 --guppy-server-path /usr/bin/guppy_basecall_server
WARNING! The BED file produced by megalodon is not sorted – if you use it for downstream analysis it will give inaccurate results. To sort:
bedtools sort -i modified_bases.5mC.bed > modified_bases.5mC.sorted.bed
Column 10 is the read depth at a particular site; column 11 is the % of those reads that had a CG modification. Most of the filtering will be done on those two metrics. For example, you may want to remove sites with no coverage so they don’t overrepresent the propartion of unmethylated sites in the genome:
awk -F"\t" '{if ($10 == 0) print $0}' modified_bases.5mC.sorted.bed > modified_bases.5mC.sorted.rmMissing.bed
Comparing methylated sites and genome features also requires a GFF format file for the features of interest.
Gene Body Methylation
This section calculated the rate of methylated CG sites over gene exons and adjacent upstream/downstream non-coding regions. Some important caveats for my approach:
- Only hornwort annotations produced by BRAKER have been tested
- Some field values (esp. GFF column 3) may be different in other genomes
- "gene" objects are expected to represent only the gene body between start and stop codons (inclusive), not UTRs. On gene lines in the GFF file, field 9 must be ID=geneID, where geneID is whatever unique identifier they have. The gene ID cannot be a substring of another element in the annotation file. E.g. genes labeled g10 and g100 cannot be used (otherwise grep commands will find the wrong matches)
- "exon" objects are expected to represent just the parts that make up the CDS. They must include the parent gene ID somewhere in field 9
First, some setup:
# Make a list of gene IDs, used for searching later
awk -F"\t" '{ if ($3 == "gene") print $9}' gene_annotations.gff | cut -f 2 -d "=" >> geneIDs.txt
# Make subdirectories for new files
mkdir genes
mkdir exons
mkdir upstream_1kb
mkdir downstream_1kb
Next, get just the “gene” lines out of the GFF, making a new file for each gene in the ./genes/ directory:
cat geneIDs.txt | while read i ; do grep "$i" gene_annotations.gff | awk -F"\t" '{ if ($3 == "gene") print $0}' > genes/"$i".gff ; done
Get just the exon lines from the main GFF, put into one new file in the ./exons directory:
awk -F"\t" '{ if ($3 == "exon") print $0}' gene_annotations.gff | sortBed -i - | bedtools merge -i - > exons/exons.bed
Calculate the upstream and downstream 1kb regions for each gene. The long awk command simply looks at the orientation of the gene, then adds or subtracts 1000 to the gene region as appropriate, and formats the new region in GFF format.
awk -F"\t" '{ if ($3 == "gene" && $7 == "+") print $1"\t"$2"\tupstream\t"($4-1000)"\t"$4"\t"$6"\t"$7"\t"$8"\t"$9; else if ($3 =="gene" && $7 == "-" ) print ($1"\t"$2"\tupstream\t"$5"\t"($5+1000)"\t"$6"\t"$7"\t"$8"\t"$9) }' gene_annotations.gff > upstream_1kb/upstream.gff
awk -F"\t" '{ if ($3 == "gene" && $7 == "+") print $1"\t"$2"\tdownstream\t"$5"\t"($5+1000)"\t"$6"\t"$7"\t"$8"\t"$9; else if ($3 == "gene" && $7 == "-" ) print ($1"\t"$2"\tdownstream\t"$4-1000"\t"$4"\t"$6"\t"$7"\t"$8"\t"$9) }' gene_annotations.gff > downstream_1kb/downstream.gff
Remove any negative values from the upstream/downstream regions (for genes near ends of sequences):
awk -F"\t" '{ if ($4 < 1) print $1"\t"$2"\t"$3"\t1\t"$5"\t"$6"\t"$7"\t"$8"\t"$9; else print $0}' upstream_1kb/upstream.gff > upstream_1kb/upstream.pos.gff
awk -F"\t" '{ if ($4 < 1) print $1"\t"$2"\t"$3"\t1\t"$5"\t"$6"\t"$7"\t"$8"\t"$9; else print $0}' downstream_1kb/downstream.gff > downstream_1kb/downstream.pos.gff
Now that all the gene region files are ready, we can start to intersect those regions with the 5mC CG modifications BED file:
bedtools intersect -a modified_bases.5mC.sorted.bed -b exons/exons.bed > exons/exons.CpG.bed
bedtools intersect -a modified_bases.5mC.sorted.bed -b upstream_1kb/upstream.pos.gff > upstream_1kb/upstream.CpG.bed
bedtools intersect -a modified_bases.5mC.sorted.bed -b downstream_1kb/downstream.pos.gff > downstream_1kb/downstream.CpG.bed
Since sites with low coverage may innaccurately represent their true % modification, you may want to remove them. The cutoff will vary depending on your experiment and sequencing depth. This example requires at least 5 reads per site, in a dataset with median sequencing depth of ~50X.
awk -F"\t" '{if ($10 >= 5) print $1"\t"$2"\t"$3"\t"$4"\t"$11}' exons/exons.CpG.bed > exons/exons.CpG.filt.bed
awk -F"\t" '{if ($10 >= 5) print $1"\t"$2"\t"$3"\t"$4"\t"$11}' upstream_1kb/upstream.CpG.bed > upstream_1kb/upstream.CpG.filt.bed
awk -F"\t" '{if ($10 >= 5) print $1"\t"$2"\t"$3"\t"$4"\t"$11}' downstream_1kb/downstream.CpG.bed > downstream_1kb/downstream.CpG.filt.bed
Next, the CG modifications are averaged over portions of each sequence. In upstream/downstream regions, 100 bp windows are used (-n 10); since gene lengths vary, each gene is broken into 20 windows (-n 20) that will vary in size between genes. Having an equal number of windows is necessary to be able to “stack” all the genes for comparison.
WARNING: These steps are slow (at least several minutes). Do within exons, upstream_1kb, and downstream_1kb directories:
# For upstream/downstream:
cat ../geneIDs.txt | while read i ; do grep "$i" upstream.pos.gff | bedtools makewindows -n 10 -b - | sortBed -i - | bedmap --echo --mean - upstream.CpG.filt.bed | sed -e 's/|/\t/g' | awk -F"\t" '{print $1"\t"$2"\t"$3"\t"$4}' > "$i".windowavg.bed ; done
cat ../geneIDs.txt | while read i ; do grep "$i" downstream.pos.gff | bedtools makewindows -n 10 -b - | sortBed -i - | bedmap --echo --mean - downstream.CpG.filt.bed | sed -e 's/|/\t/g' | awk -F"\t" '{print $1"\t"$2"\t"$3"\t"$4}' > "$i".windowavg.bed ; done
# For exons:
cat ../geneIDs.txt | while read i ; do bedtools makewindows -n 20 -b ../genes/"$i".gff | sortBed -i - | bedmap --echo --mean - exons.CpG.filt.bed | sed -e 's/|/\t/g' | awk -F"\t" '{print $1"\t"$2"\t"$3"\t"$4}' > "$i".windowavg.bed ; done
Reorient genes so they’re all in the same direction. Repeat in each upsteam, downstream, exons directory.
cat ../geneIDs.txt | while read i ; do grep "$i" ../gene_annotations.gff | grep -w "gene" | if [ $(cut -f 7) == "+" ] ; then cat "$i".windowavg.bed > "$i".windowavg.oriented.bed ; else tac "$i".windowavg.bed > "$i".windowavg.oriented.bed; fi ; done
Remove any genes that were too short or pseudogenes and did not produce output:
find . -size 0 -delete
Convert everything into long-data format to send to R for plotting. Do this in the directory above the separate exons, upstream, and downstream ones.
for i in upstream_1kb/*oriented.bed ; do awk -F"\t" -v j=1 '{print j++"\t"$4}' "$i" >> combined.tsv ; done
for i in exons/*oriented.bed ; do awk -F"\t" -v j=11 '{print j++"\t"$4}' "$i" >> combined.tsv ; done
for i in downstream_1kb/*oriented.bed ; do awk -F"\t" -v j=31 '{print j++"\t"$4}' "$i" >> combined.tsv ; done
# Remove any missing data
awk -F"\t" '{ if ($2 != "NAN") print $0}' combined.tsv > combined.rmNA.tsv
Import the data into R to produce a line plot of the averaged CG methylation:
library(ggplot2)
speciesName <- "Species name"
combined <- read.delim("combined.rmNA.tsv", header = F, sep = "\t")
combined$V1 <- as.numeric(combined$V1)
ggplot(data = combined, aes(x=V1, y=V2)) +
stat_summary(fun.min = function(z) { quantile(z,0.25) },
fun.max = function(z) { quantile(z,0.75) },
fun = median, geom = "line", color = "red") +
ylab("Median % CG modification") +
xlab("") +
ylim(0,100) +
ggtitle(speciesName) +
theme_bw() +
scale_x_continuous(breaks = c(1,11,30,40), labels=c("1" = "-1 Kbp", "11" = "Start", "30" = "Stop", "40" = "+1 Kbp"))
Orthogroup InferenceTop
Orthogroups were constructed with Orthofinder 2.5.4 using protein sequence input from genomes selected to represent a broad sampling across Viridiplantae. Because each genome and its genes are annotated differently, custom filtering methods were needed for most genomes to create informative sequence IDs and remove alternative transcripts. A selection of methods follow:
Isoetes taiwanensis: restructure names to represent just the unique transcript ID and remove alternative transcripts.
grep ">" Isoetes_taiwanensis_61511-CDS-prot.fasta | awk -F"|" '{print $0"\t"$9}' > Isoetes_taiwanensis_61511-CDS-prot.fasta.rename-table.tsv
sed -i "s/>//" Isoetes_taiwanensis_61511-CDS-prot.fasta.rename-table.tsv
renameFasta.py Isoetes_taiwanensis_61511-CDS-prot.fasta Isoetes_taiwanensis_61511-CDS-prot.fasta.rename-table.tsv
grep "\-RA" Isoetes_taiwanensis_61511-CDS-prot_renamed.fasta | getFromFasta.py Isoetes_taiwanensis_61511-CDS-prot_renamed.fasta - >
Isoetes_taiwanensis_61511-CDS-prot_renamed.primary_transcripts.fasta
Chara braunii: remove alternative (shortest) transcripts when sequences are named with the .t<number> suffix.
removeAlternativeTranscripts.py chbra_iso_noTE_23546_pep.fasta
Alsophila spinulosa: replace in-frame stop codons with unknown amino acids. In Python:
import re
with open("Alsophila_spinulosa_v3.1_protein.fa", "r") as infile, open("Alsophila_spinulosa_v3.1_protein.IFSconverted.fa", "w") as outfile:
for line in infile:
if line.startswith(">"):
outfile.write(line)
else:
outfile.write(re.sub("\.", "X", line))
Spirogloea muscicola: check for alternative transcripts when lacking any information in transcript id:
# Convert FASTA header information to BED format:
grep ">" Spirogloea_muscicola_gene.pep.fasta | awk -F" |:|=" '{print $6"\t"$7"\t"$8"\t"$1"\t0\t"$9}' | sort -k 1,1 -k 2,2n > Spirogloea_muscicola_gene.pep.fasta.sorted.bed
# Use bedtools to merge overlapping or touching regions:
bedtools merge -i Spirogloea_muscicola_gene.pep.fasta.sorted.bed -d 0 -s -c 1,4 -o count,distinct | awk -F"\t" '{ if ( $4 > 1 ) print $0}'
Once all genomes were checked and modified as needed, run Orthofinder:
orthofinder -M msa -t 24 -S diamond -A mafft -T fasttree -X -f ./orthofinder_input/
With the sequence files for each orthogroup, I generated more accurate phylogenies by aligning each orthogroup with Clustal Omega, trimming sites with >90% gaps to remove spurious alignments, and inferred phylognies with IQ-TREE with standard model selection and 5000 ultrafast bootstrap replicates. Note use of find because the file list is usually too long for using wildcards.
find . -name "OG*fa" | while read i ; do clustalo --auto -i "$i" -o "$i".CLUSTAL.fa ; done
find . -name "OG*CLUSTAL.fa" | while read i ; do trimal -in "$i" -out "$i".TRIM.fa -gt 0.1
find . -name "OG*TRIM.fa" | while read i ; do iqtree -s "$i" -T 4 -B 5000 -m TEST ; done
SyntenyTop
Extracting syntenic blocks
Synteny among hornwort genomes was inferred with GENESPACE. I attempted to run with other bryophyte genomes, but no synteny was found.
Primary transcript protein FASTAs were downloaded from HornwortBase, Phytozome, CoGe, and other sources. Because naming scemes across GFF/FASTA pairs varies, each genome had to be manually inspected and different methods developed to match the sequence names in the FASTA with the GFF entries. GFFs then need to be subsetted so there is a single entry per sequence in the FASTA. Finally, GFF files were converted into a simplified 4-column BED format, and FASTA and BED files were renamed to match each other.
Convert Phytozome FASTAs
grep ">" CpurpureusGG1_539_v1.1.protein_primaryTranscriptOnly.fa | awk -F" |=" '{print $0"\t"$7}' > Ceratodon_purpureus_GG1.convert.tsv
renameFasta.py CpurpureusGG1_539_v1.1.protein_primaryTranscriptOnly.fa Ceratodon_purpureus_GG1.convert.tsv
mv CpurpureusGG1_539_v1.1.protein_primaryTranscriptOnly_renamed.fa Ceratodon_purpureus_GG1.fa
Convert GFF to simplified BED
For Phytozome:
grep -w "gene" Ppatens_318_v3.3.gene.gff3 | gff2bed | awk -F"\t|=|;" '{print $1"\t"$2"\t"$3"\t"$13}' > Ppatens_318_v3.3.gene.bed
For HornwortBase:
grep -w "gene" Anthoceros_agrestis_Oxford_gene_annotations.gff | gff2bed | awk -F"\t|=|;" '{print $1"\t"$2"\t"$3"\t"$11}' > Anthoceros_agrestis_Oxford.bed
Run Genespace in R:
library(GENESPACE)
gpar <- init_genespace(wd = "~/genespace", path2mcscanx = "~/bin/MCScanX/"
out <- run_genespace(gsParam = gpar)
Information about syntenic blocks was extracted from GENESPACE output file in GENESPACE-DIR/results/syntenicBlock_coordinates.tsv.
To get stats about block size between pairs of genomes:
grep "AnagrBONN" syntenicBlock_coordinates.csv | grep "AnagrOXF" | sed 's/,/\t/g' | cut -f 5,12 | sort | uniq | cut -f 2 | summaryStats.R
Note that the sort and uniq commands are used to avoid double-counting the same block in both orientations.
Gene ExpressionTop
Gene expression was calculated by mapping RNA reads to the respective genome with Hisat2 and analyzed with Stringtie. Hisat2 indexes were built with splice site and exon information generated from the genome’s GTF annotation file. The hisat2 script hisat2_extract_exons.py was modified to work with GTF files produced by BRAKER.
hisat2_extract_splice_sites.py braker.gtf > ss.txt
hisat2_extract_exons_BRAKER.py > exon.txt
hisat2-build --ss ss.txt --exon exon.txt genome.fasta genome.fasta
Differential expression in Notothylas orbicularis followed the HISAT2-Stringtie-Ballgown pipeline as described in the Stringtie documentation (). In brief, each replicate RNAseq dataset was mapped to the genome with HISAT2 and the alignment used to assemble transcripts with Stringtie.
hisat2 -p 24 --dta -x genome.fasta -1 sample1_RNA_reads_R1.fq.gz -2 sample1_RNA_reads_R2.fq.gz | samtools sort -o genome.sample1_RNA.bam
for i in genome.sample*RNA.bam ; do stringtie -o "$i".stringtie.gtf -G braker.gtf "$i" ; done
Assemblies were merged with Stringtie to create a non-redundant set of transcripts, and then transcript abundance was estimated for each individual replicate.
stringtie --merge -o stringtie.merged.gtf -G braker.gtf *stringtie.gtf
for i in genome.sample*RNA.bam ; do stringtie -o "$i".stringtie.merged -G stringtie.merged.gtf
Statistical analysis of the abundances was performed in Ballgown ().
GO Term EnrichmentTop
GO term enricnment was used to analyse genes found on sex and accessory chromosomes. Goatools was used on GO term files created from eggNOG functional annotations. Statistical signifance cutoff was Bonferroni-corrected p =< 0.05.
First, create a table of each gene transcript and its associated GO terms. Convert semicolons into commas.
cut -f 1,10 Hornwort_functional_annotations.tsv | sed 's/;/,/g' > Hornwort_GO_table.tsv
Create two new text files with transcript IDs.
The first file is the “background” with all primary transcripts in a genome.
The second file is the “targets” with transcripts to be compared against the background.
In this example, I’m comparing an accessory chromosome to the whole genome.
It doesn’t matter if the full transcript, CDS, or protein file is used since the sequence IDs are the same.
The cut command is needed to remove FASTA metadata in the sequence ID line and the sed command to remove greater-than symbols.
grep ">" Anthoceros_agrestis_Oxford_CDS_primary_transcripts.fasta | cut -f 1 -d " " | sed 's/>//' > Anthoceros_agrestis_Oxford.background.txt
grep "AnagrOXF.S6" > Anthoceros_agrestis_Oxford.targets.txt
Run goatools with these files. If you haven’t downloaded the GO database, get it from here and include the path to it in the command.
find_enrichment.py --obo ~/Downloads/go-basic.obo --method bonferroni --outfile Anthoceros_agrestis_Oxford.S6.enrichment_results.xlsx Anthoceros_agrestis_Oxford.targets.txt Anthoceros_agrestis_Oxford.background.txt Hornwort_GO_table.tsv
Whole Genome DuplicationTop
Whole genome duplication (WGD) was inferred from Ks (synonymous substitution) plots from wgd (). The input is the CDS sequence fasta containing only primary transcripts. Self-synteny plots were visually inspected for any indication of 2-to-1 syntenic block ratios typical of WGD.
wgd dmd --nostrictcds --ignorestop Anthoceros_agrestis_Oxford_CDS_primary_transcripts.fasta
wgd ksd --n_threads 24 Anthoceros_agrestis_Oxford_CDS_primary_transcripts.fasta.mcl Anthoceros_agrestis_Oxford_CDS_primary_transcripts.fasta
wgd syn -f gene -a ID -ks Anthoceros_agrestis_Oxford_CDS_primary_transcripts.fasta.tsv Anthoceros_agrestis_Oxford_gene_annotations.gff Anthoceros_agrestis_Oxford_CDS_primary_transcripts.fasta.mcl
Sex Chromosome MappingTop
Sex chromosomes were inferred by mapping short-read data from 5-6 individuals of each species to the respective reference genome. Data were first filtered to remove organellar reads (which represented up to 50% of raw data) and bacterial contaminants if present, remove PCR duplicates, and then randomly sampled to an equal amount of data (in Mbp). Reads were mapped to the genome and median depth was calculated in each 100 kbp window across the genome. For each window, the depth for each individual was subracted from the control (data from the individual used to assemble the genome).
Map reads to the organelles, remove PCR duplicates, then keep reads that do not map and save them as FASTQ files. Repeat for each individual.
cat Species_plastome.fasta Species_chondrome.fasta > Species_organelles.fasta
bwa index Species_organelles.fasta
bwa mem -t 24 Species_organelles.fasta Species_individualA_R1.fq.gz Species_individualA_R2.fq.gz | samblaster | samtools view -f 4 | samtools sort -n | samtools fastq -1 Species_individualA_organellesFiltered_R1.fq -2 Species_individualA_organellesFiltered_R2.fq
Subset the reads of the control individual to match each other individual (since the control has the most data to start). First find the amount of data for each. I’m using the assembly-stats script but you can use anything that reports the total amount of data and number of reads.
assembly-stats -t *organellesFiltered*.fq
Use seqtk to randomly sample from the read files. Seqtk operates on number of reads, but having the same total amount of data is more important for depth-based analyses. You can start sampling using the number of reads, but it may need to be adjusted to get the amount of data more even. If you know the read length distribution for each dataset you can use it to calculate the number of reads to reach a certain amount of data. Repeat for each sample being compared to the control.
seqtk sample -s100 Species_controlIndividual_organellesFiltered_R1.fq 14613074 > Species_controlIndividual_subset_to_individualA_R1.fq
seqtk sample -s100 Species_controlIndividual_organellesFiltered_R2.fq 14613074 > Species_controlIndividual_subset_to_individualA_R2.fq
Map reads from each individual, including the control, to the reference genome.
bwa index Species_genome.fasta
bwa mem -t 24 Species_genome.fasta Species_individualA_R1.fq Species_individualA_R2.fq | samtools sort -o Species_individualA.bam
Get the read depth across the genome for each individual.
samtools depth Species_individualA.bam > Species_individualA.depth.txt
Convert the samtools depth output into BED format.
awk -F"\t" '{print $1"\t"$2"\t"$2+1"\t"$3"\t"$3}' Species_individualA.depth.txt > Species_individualA.depth.bed
Make a reference BED file for the genome. Used for making windows for calculations.
samtools faidx Species_genome.fasta
awk -F"\t" '{print $1"\t1\t"$2}' Species_genome.fasta.fai > Species_genome.bed
Calculate the median depth value in each 100kbp window. Repeat for each individual and each subset of the control subsampled to the same amount of data as each other individual.
bedtools makewindows -b Species_genome.bed -w 100000 | sortBed -i - | bedmap --echo --median - Species_individualA.depth.bed | sed 's/|/\t/g' > Species_individualA.depth.100kb.bed
The next steps are done in R.
A few packages that are required are ggplot2, ggpubr, dplyr, and superb.
With those installed, read in your data files. I’ve grouped these by the separate individuals vs. control individual.
This example uses Phymatoceros phymatodes from Fig. 4.
library(ggplot2)
library(ggpubr)
library(superb)
library(dplyr)
# read files and set up dataframes
Onedepth <- read.delim("PhymatoCJR5469_1.genomeMapped.bam.depth.100kb.bed", header = F, sep = "\t")
Twodepth <- read.delim("PhymatoCJR5469_2.genomeMapped.bam.depth.100kb.bed", header = F, sep = "\t")
Threedepth <- read.delim("PhymatoCJR5469_3.genomeMapped.bam.depth.100kb.bed", header = F, sep = "\t")
Fivedepth <- read.delim("PhymatoCJR5469_5.genomeMapped.bam.depth.100kb.bed", header = F, sep = "\t")
Sixdepth <- read.delim("PhymatoCJR5469_6.genomeMapped.bam.depth.100kb.bed", header = F, sep = "\t")
HsubsetOnedepth <- read.delim("Phymatoceros_phymatodes_H40.subset1.genomeMapped.bam.depth.100kb.bed", header = F, sep = "\t")
HsubsetTwodepth <- read.delim("Phymatoceros_phymatodes_H40.subset2.genomeMapped.bam.depth.100kb.bed", header = F, sep = "\t")
HsubsetThreedepth <- read.delim("Phymatoceros_phymatodes_H40.subset3.genomeMapped.bam.depth.100kb.bed", header = F, sep = "\t")
HsubsetFivedepth <- read.delim("Phymatoceros_phymatodes_H40.subset5.genomeMapped.bam.depth.100kb.bed", header = F, sep = "\t")
HsubsetSixdepth <- read.delim("Phymatoceros_phymatodes_H40.subset6.genomeMapped.bam.depth.100kb.bed", header = F, sep = "\t")
colnames(Onedepth) <- c("SampleChrom","SampleBase", "StopBase", "SampleDepth")
colnames(Twodepth) <- c("SampleChrom","SampleBase", "StopBase", "SampleDepth")
colnames(Threedepth) <- c("SampleChrom","SampleBase", "StopBase", "SampleDepth")
colnames(Fivedepth) <- c("SampleChrom","SampleBase", "StopBase", "SampleDepth")
colnames(Sixdepth) <- c("SampleChrom","SampleBase", "StopBase", "SampleDepth")
colnames(HsubsetOnedepth) <- c("RefChrom","RefBase", "RefStop", "RefDepth")
colnames(HsubsetTwodepth) <- c("RefChrom","RefBase", "RefStop", "RefDepth")
colnames(HsubsetThreedepth) <- c("RefChrom","RefBase", "RefStop", "RefDepth")
colnames(HsubsetFivedepth) <- c("RefChrom","RefBase", "RefStop", "RefDepth")
colnames(HsubsetSixdepth) <- c("RefChrom","RefBase", "RefStop", "RefDepth")
Onedepth$SampleDepth <- as.numeric(Onedepth$SampleDepth)
Twodepth$SampleDepth <- as.numeric(Twodepth$SampleDepth)
Threedepth$SampleDepth <- as.numeric(Threedepth$SampleDepth)
Fivedepth$SampleDepth <- as.numeric(Fivedepth$SampleDepth)
Sixdepth$SampleDepth <- as.numeric(Sixdepth$SampleDepth)
HsubsetOnedepth$RefDepth <- as.numeric(HsubsetOnedepth$RefDepth)
HsubsetTwodepth$RefDepth <- as.numeric(HsubsetTwodepth$RefDepth)
HsubsetThreedepth$RefDepth <- as.numeric(HsubsetThreedepth$RefDepth)
HsubsetFivedepth$RefDepth <- as.numeric(HsubsetFivedepth$RefDepth)
HsubsetSixdepth$RefDepth <- as.numeric(HsubsetSixdepth$RefDepth)
Now combine the respective sample individual and its control.
Onemerged <- cbind(Onedepth, HsubsetOnedepth)
Twomerged <- cbind(Twodepth, HsubsetTwodepth)
Threemerged <- cbind(Threedepth, HsubsetThreedepth)
Fivemerged <- cbind(Fivedepth, HsubsetFivedepth)
Sixmerged <- cbind(Sixdepth, HsubsetSixdepth)
Remove entries where the control is missing data, has no coverage, or has much higher coverage than expected under a normal distribution (>100X).
Onemerged_rmNA <- Onemerged[Onemerged$RefDepth != "NaN", ]
Twomerged_rmNA <- Twomerged[Twomerged$RefDepth != "NaN", ]
Threemerged_rmNA <- Threemerged[Threemerged$RefDepth != "NaN", ]
Fivemerged_rmNA <- Fivemerged[Fivemerged$RefDepth != "NaN", ]
Sixmerged_rmNA <- Sixmerged[Sixmerged$RefDepth != "NaN", ]
Onemerged_rmmissing <- Onemerged_rmNA[Onemerged_rmNA$RefDepth != 0.0, ]
Twomerged_rmmissing <- Twomerged_rmNA[Twomerged_rmNA$RefDepth != 0.0, ]
Threemerged_rmmissing <- Threemerged_rmNA[Threemerged_rmNA$RefDepth != 0.0, ]
Fivemerged_rmmissing <- Fivemerged_rmNA[Fivemerged_rmNA$RefDepth != 0.0, ]
Sixmerged_rmmissing <- Sixmerged_rmNA[Sixmerged_rmNA$RefDepth != 0.0, ]
Onemerged_rmOutliers <- Onemerged_rmmissing[which((Onemerged_rmmissing$SampleDepth < 100 ) & (Onemerged_rmmissing$RefDepth < 100)), ]
Twomerged_rmOutliers <- Twomerged_rmmissing[which((Twomerged_rmmissing$SampleDepth < 100 ) & (Twomerged_rmmissing$RefDepth < 100)), ]
Threemerged_rmOutliers <- Threemerged_rmmissing[which((Threemerged_rmmissing$SampleDepth < 100 ) & (Threemerged_rmmissing$RefDepth < 100)), ]
Fivemerged_rmOutliers <- Fivemerged_rmmissing[which((Fivemerged_rmmissing$SampleDepth < 100 ) & (Fivemerged_rmmissing$RefDepth < 100)), ]
Sixmerged_rmOutliers <- Sixmerged_rmmissing[which((Sixmerged_rmmissing$SampleDepth < 100 ) & (Sixmerged_rmmissing$RefDepth < 100)), ]
Calulate the net depth in each window by subtracting the control from the sample.
OnenetDepth <- data.frame(Chrom = Onemerged_rmOutliers$SampleChrom, Base = Onemerged_rmOutliers$SampleBase, NetDepth = (Onemerged_rmOutliers$SampleDepth - Onemerged_rmOutliers$RefDepth))
TwonetDepth <- data.frame(Chrom = Twomerged_rmOutliers$SampleChrom, Base = Twomerged_rmOutliers$SampleBase, NetDepth = (Twomerged_rmOutliers$SampleDepth - Twomerged_rmOutliers$RefDepth))
ThreenetDepth <- data.frame(Chrom = Threemerged_rmOutliers$SampleChrom, Base = Threemerged_rmOutliers$SampleBase, NetDepth = (Threemerged_rmOutliers$SampleDepth - Threemerged_rmOutliers$RefDepth))
FivenetDepth <- data.frame(Chrom = Fivemerged_rmOutliers$SampleChrom, Base = Fivemerged_rmOutliers$SampleBase, NetDepth = (Fivemerged_rmOutliers$SampleDepth - Fivemerged_rmOutliers$RefDepth))
SixnetDepth <- data.frame(Chrom = Sixmerged_rmOutliers$SampleChrom, Base = Sixmerged_rmOutliers$SampleBase, NetDepth = (Sixmerged_rmOutliers$SampleDepth - Sixmerged_rmOutliers$RefDepth))
Extract just the data for the chromosome-scale scaffolds (ignore the unscaffolded contigs). Add a numerical identifier and rename the columns.
OnenetDepthScaff <- subset(OnenetDepth, OnenetDepth$Chrom %in% c("Phphy.S1","Phphy.S2","Phphy.S3","Phphy.S4","Phphy.S5"))
TwonetDepthScaff <- subset(TwonetDepth, TwonetDepth$Chrom %in% c("Phphy.S1","Phphy.S2","Phphy.S3","Phphy.S4","Phphy.S5"))
ThreenetDepthScaff <- subset(ThreenetDepth, ThreenetDepth$Chrom %in% c("Phphy.S1","Phphy.S2","Phphy.S3","Phphy.S4","Phphy.S5"))
FivenetDepthScaff <- subset(FivenetDepth, FivenetDepth$Chrom %in% c("Phphy.S1","Phphy.S2","Phphy.S3","Phphy.S4","Phphy.S5"))
SixnetDepthScaff <- subset(SixnetDepth, SixnetDepth$Chrom %in% c("Phphy.S1","Phphy.S2","Phphy.S3","Phphy.S4","Phphy.S5"))
OnenetDepthFinal <- cbind("1", OnenetDepthScaff)
TwonetDepthFinal <- cbind("2", TwonetDepthScaff)
ThreenetDepthFinal <- cbind("3", ThreenetDepthScaff)
FivenetDepthFinal <- cbind("5", FivenetDepthScaff)
SixnetDepthFinal <- cbind("6", SixnetDepthScaff)
colnames(OnenetDepthFinal) <- c("Sample", "Chrom", "Base", "NormDepth")
colnames(TwonetDepthFinal) <- c("Sample", "Chrom", "Base", "NormDepth")
colnames(ThreenetDepthFinal) <- c("Sample", "Chrom", "Base", "NormDepth")
colnames(FivenetDepthFinal) <- c("Sample", "Chrom", "Base", "NormDepth")
colnames(SixnetDepthFinal) <- c("Sample", "Chrom", "Base", "NormDepth")
Combine individual dataframes together.
combined <- bind_rows(OnenetDepthFinal, ThreenetDepthFinal, TwonetDepthFinal, FivenetDepthFinal, SixnetDepthFinal)
combined$Sample <- factor(combined$Sample, levels = c("1", "3", "2", "5", "6"))
Plot the net depth using boxplots for each chromosome and individual.
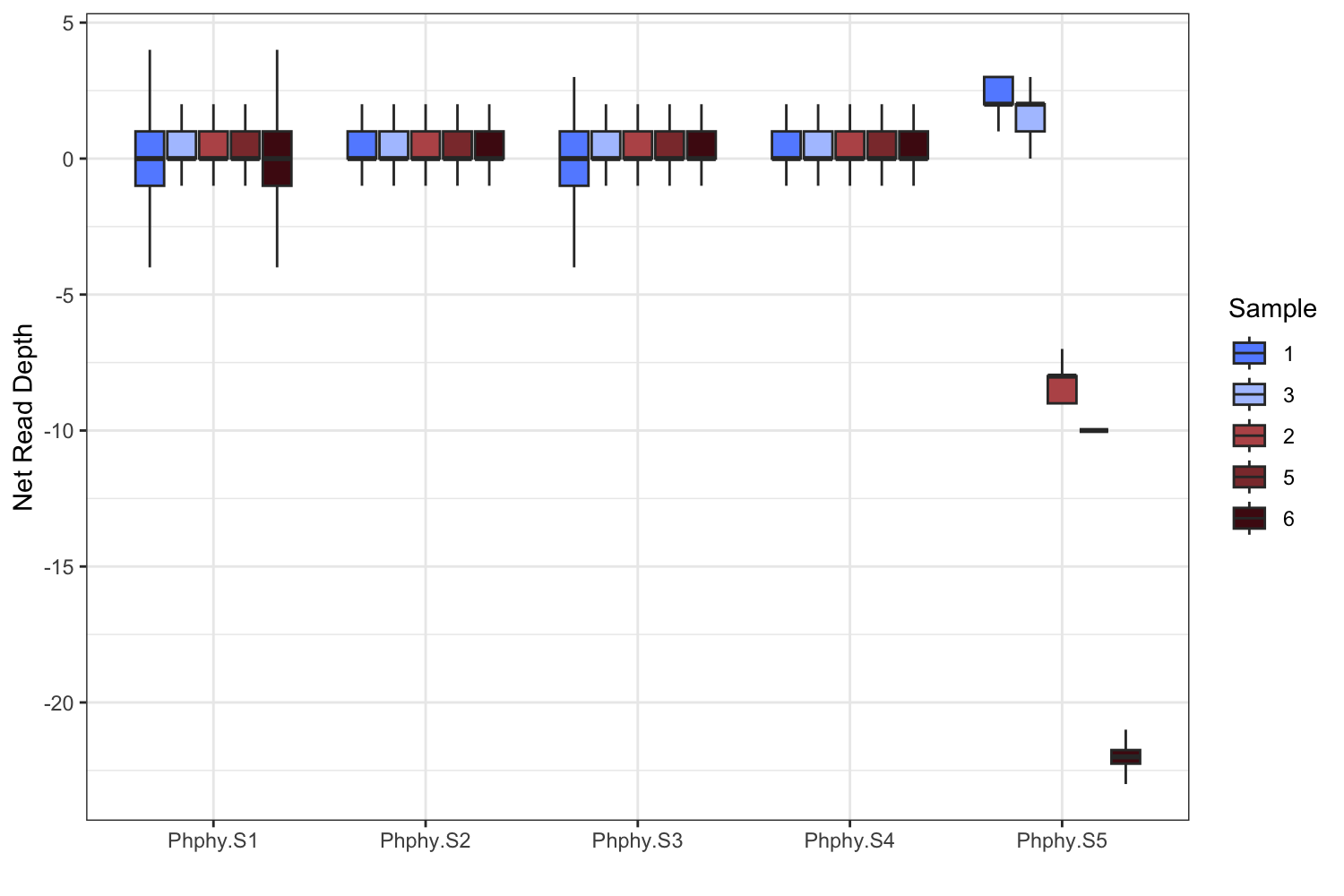
ggplot(data = combined, aes(x = Chrom, y = NormDepth, fill = Sample)) +
geom_boxplot(outliers = F, outlier.alpha = 0.1, outlier.shape = ".") +
#ylim(0.35, 0.65) +
xlab("") +
scale_fill_manual(values=c("#648FFF", "#AFC5FF", "#b95657", "#8c383a", "#4e0d14")) +
ylab("Net Read Depth") +
theme_bw()
You should have something that looks like the Fig. 4c left panel.
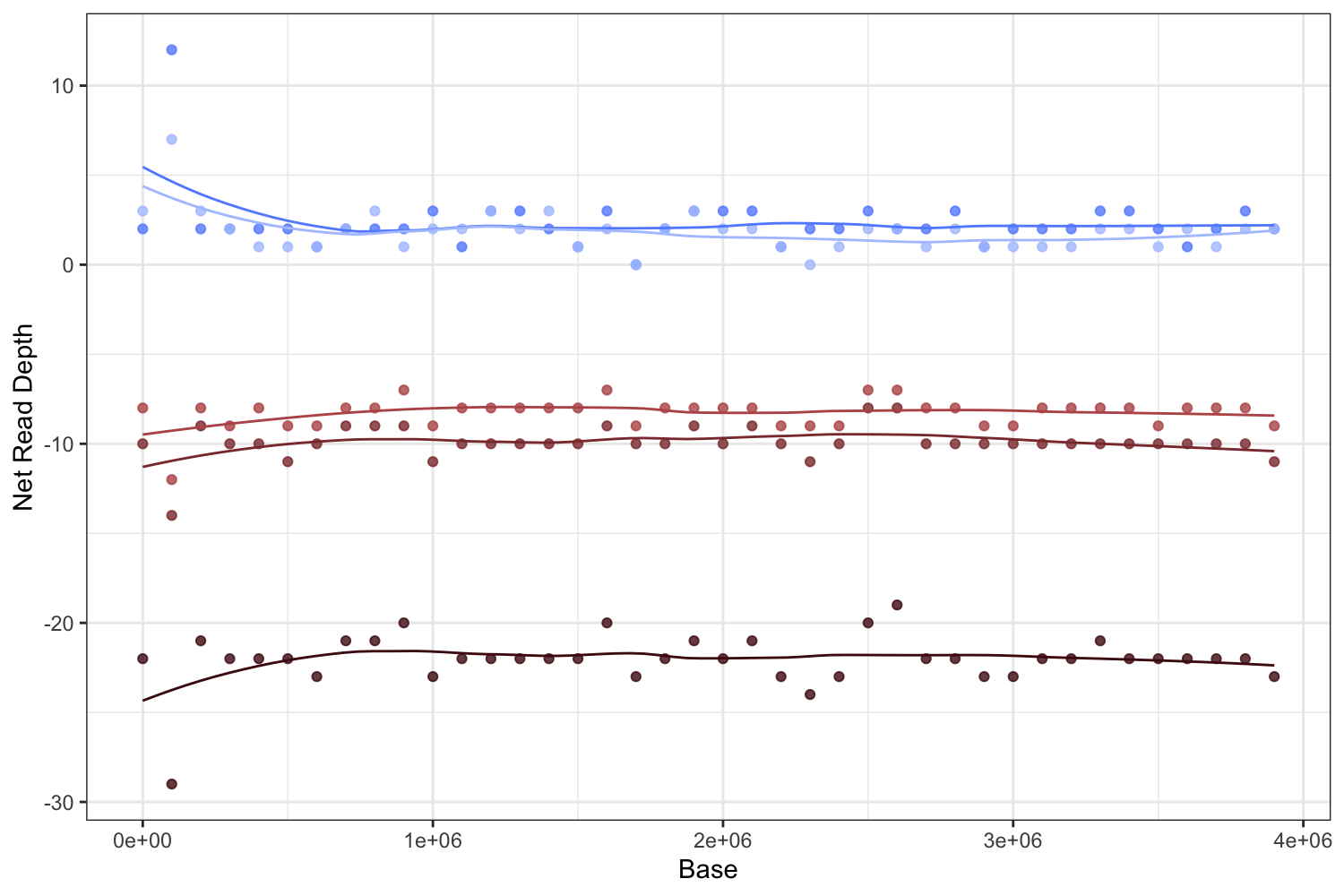
Now extract the data just for the sex chromosome so we can plot the net depth along the chromosome for each individual.
One.S5.netDepth <- subset(OnenetDepthFinal, OnenetDepthFinal$Chrom %in% "Phphy.S5")
Two.S5.netDepth <- subset(TwonetDepthFinal, TwonetDepthFinal$Chrom %in% "Phphy.S5")
Three.S5.netDepth <- subset(ThreenetDepthFinal, ThreenetDepthFinal$Chrom %in% "Phphy.S5")
Five.S5.netDepth <- subset(FivenetDepthFinal, FivenetDepthFinal$Chrom %in% "Phphy.S5")
Six.S5.netDepth <- subset(SixnetDepthFinal, SixnetDepthFinal$Chrom %in% "Phphy.S5")
Combine into a single dataframe.
combinedS5malefemale <- bind_rows(One.S5.netDepth, Three.S5.netDepth,Two.S5.netDepth, Five.S5.netDepth, Six.S5.netDepth )
Plot the net depth using a dotplot with LOESS smoothed lines.
ggplot(data = combinedS5malefemale, aes(x = Base, y = NormDepth, color = Sample)) +
geom_point(alpha = 0.8, show.legend = F) +
geom_line(show.legend = F, stat = "smooth", span = 0.5) +
ylab("Net Read Depth") +
scale_color_manual(values=c("#648FFF", "#b95657", "#AFC5FF", "#8c383a", "#4e0d14")) +
theme_bw()
The result is the Fig 4c. right panel.
Pairwise Wilcoxon rank sum tests can be used to test the significance of the difference in depth between sex chromosome and autosomes, with Bonferroni correction for multiple comparisons.
pairwise.wilcox.test(OnenetDepthFinal$NormDepth, OnenetDepthFinal$Chrom, p.adjust.method = "bonf")
pairwise.wilcox.test(TwonetDepthFinal$NormDepth, TwonetDepthFinal$Chrom, p.adjust.method = "bonf")
pairwise.wilcox.test(ThreenetDepthFinal$NormDepth, ThreenetDepthFinal$Chrom, p.adjust.method = "bonf")
pairwise.wilcox.test(FivenetDepthFinal$NormDepth, FivenetDepthFinal$Chrom, p.adjust.method = "bonf")
pairwise.wilcox.test(SixnetDepthFinal$NormDepth, SixnetDepthFinal$Chrom, p.adjust.method = "bonf")
Output:
Pairwise comparisons using Wilcoxon rank sum test with continuity correction
data: OnenetDepthFinal$NormDepth and OnenetDepthFinal$Chrom
Phphy.S1 Phphy.S2 Phphy.S3 Phphy.S4
Phphy.S2 0.0104 - - -
Phphy.S3 1.0000 1.0000 - -
Phphy.S4 0.0013 1.0000 0.2558 -
Phphy.S5 < 2e-16 3.2e-15 < 2e-16 2.5e-13
P value adjustment method: bonferroni
Pairwise comparisons using Wilcoxon rank sum test with continuity correction
data: TwonetDepthFinal$NormDepth and TwonetDepthFinal$Chrom
Phphy.S1 Phphy.S2 Phphy.S3 Phphy.S4
Phphy.S2 0.00021 - - -
Phphy.S3 0.14290 1.00000 - -
Phphy.S4 0.00013 1.00000 0.68484 -
Phphy.S5 < 2e-16 < 2e-16 < 2e-16 < 2e-16
P value adjustment method: bonferroni
Pairwise comparisons using Wilcoxon rank sum test with continuity correction
data: ThreenetDepthFinal$NormDepth and ThreenetDepthFinal$Chrom
Phphy.S1 Phphy.S2 Phphy.S3 Phphy.S4
Phphy.S2 0.12 - - -
Phphy.S3 1.00 1.00 - -
Phphy.S4 1.00 1.00 1.00 -
Phphy.S5 7.6e-15 4.9e-14 3.2e-14 1.4e-13
P value adjustment method: bonferroni
Pairwise comparisons using Wilcoxon rank sum test with continuity correction
data: FivenetDepthFinal$NormDepth and FivenetDepthFinal$Chrom
Phphy.S1 Phphy.S2 Phphy.S3 Phphy.S4
Phphy.S2 0.00119 - - -
Phphy.S3 0.15792 1.00000 - -
Phphy.S4 0.00077 1.00000 1.00000 -
Phphy.S5 < 2e-16 < 2e-16 < 2e-16 < 2e-16
P value adjustment method: bonferroni
Pairwise comparisons using Wilcoxon rank sum test with continuity correction
data: SixnetDepthFinal$NormDepth and SixnetDepthFinal$Chrom
Phphy.S1 Phphy.S2 Phphy.S3 Phphy.S4
Phphy.S2 0.00017 - - -
Phphy.S3 0.00560 1.00000 - -
Phphy.S4 0.00031 1.00000 1.00000 -
Phphy.S5 < 2e-16 < 2e-16 < 2e-16 < 2e-16
P value adjustment method: bonferroni
We observe significantly decreased coverage in just one chromosome, Phphy.S5, as expected if this is the sex chromosome.
PangenomeTop
The hornwort phylum-level pangenome classified gene orthogroups based on presence/absence across the phylogeny into traditional pangenome categories of “core” (present in every genome), “peripheral” (present in all but one genome), “dispensable” (present in two to nine genomes), and “private” (present in only one genome). Results were determined from the OrthoFinder analysis of hornwort genomes, with Anthoceros agrestis Bonn and A. angustus removed due to their different gene prediction methods.
The OrthoFinder Orthogroups.GeneCount.tsv table was first stripped of the ‘Total’ column, then converted into a presence/absence table with values of 1/0, respectively.
cut -f 1-12 Orthogroups.GeneCount.tsv > Orthogroups.GeneCount.noTotal.tsv
In Python:
with open("Orthogroups.GeneCount.noTotal.tsv", "r") as infile, open("Orthogroups.PresenceAbsence.tsv", "w") as outfile:
linecount = 0
for line in infile:
linecount +=1
if linecount > 1:
splitline = line.strip("\n").split("\t")
newline = [splitline[0]]
for item in splitline[1:]:
if int(item) > 0:
newline.append("1")
else:
newline.append("0")
outfile.write("%s\n" % "\t".join(newline))
else:
outfile.write(line)
Separate orthogroups into categories based on the presence/absence table in Python:
with open("Orthogroups.PresenceAbsence.tsv", "r") as infile, \
open("Orthogroups.GeneCount.noTotal.tsv","r") as genecountfile, \
open("Orthogroups.Core.tsv", "w") as coreOutfile, \
open("Orthogroups.Peripheral.tsv","w") as periOutfile, \
open("Orthogroups.Dispensable.tsv","w") as dispOutfile, \
open("Orthogroups.Private.tsv","w") as privOutfile:
# make the dictionary of gene counts to reference later
genecountDict = {}
for line in genecountfile:
key = line.split("\t")[0]
genecountDict[key] = line
# parse the presence/absence file and sort orthogroups into categories
linecount = 0
for line in infile:
linecount += 1
if linecount > 1:
splitline = line.strip("\n").split("\t")
og = splitline[0]
numbers = [ int(x) for x in splitline[1:] ]
if sum(numbers) == 11:
coreOutfile.write(genecountDict[og])
elif sum(numbers) == 10:
periOutfile.write(genecountDict[og])
elif sum(numbers) == 1:
privOutfile.write(genecountDict[og])
else:
dispOutfile.write(genecountDict[og])
else:
coreOutfile.write(line)
periOutfile.write(line)
dispOutfile.write(line)
privOutfile.write(line)
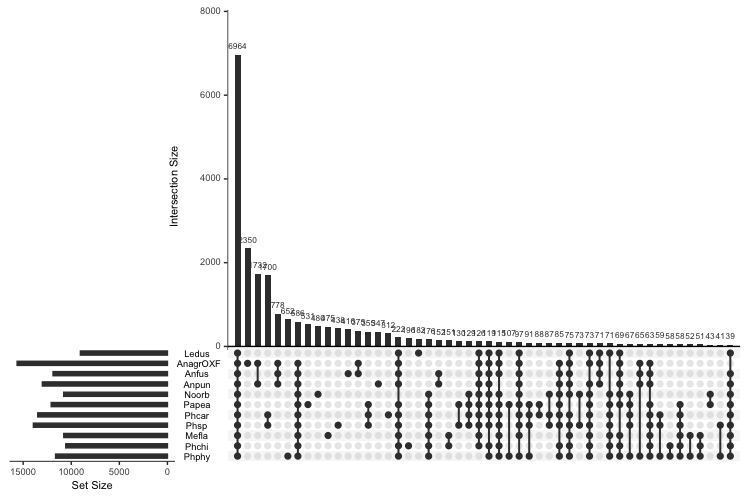
Plotting data was done in R. First, the UpSet plot:
library(UpSetR)
paDF <- read.delim("Orthogroups.PresenceAbsence.tsv", header = T, sep = "\t")
colnames(paDF) <- c("Orthogroup","AnagrOXF", "Anfus", "Anpun", "Ledus", "Mefla", "Noorb", "Papea", "Phcar", "Phsp", "Phchi", "Phphy")
upset(paDF, nsets = 14, nintersects = 50, order.by = "freq", keep.order = T, sets = rev(c("Ledus", "AnagrOXF", "Anfus", "Anpun", "Noorb", "Papea", "Phcar", "Phsp", "Mefla", "Phchi", "Phphy")))
upset(paDF, nsets = 14, nintersects = 12, order.by = "degree", keep.order = T, sets = rev(c("Ledus", "AnagrOXF", "Anfus", "Anpun", "Noorb", "Papea", "Phcar", "Phsp", "Mefla", "Phchi", "Phphy")))
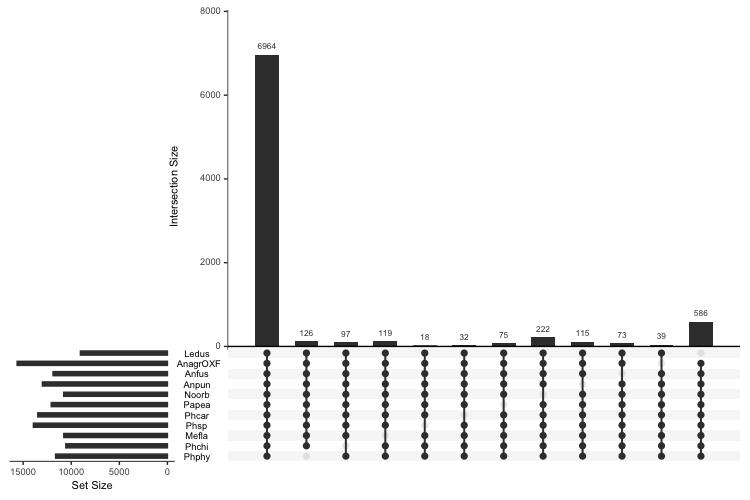
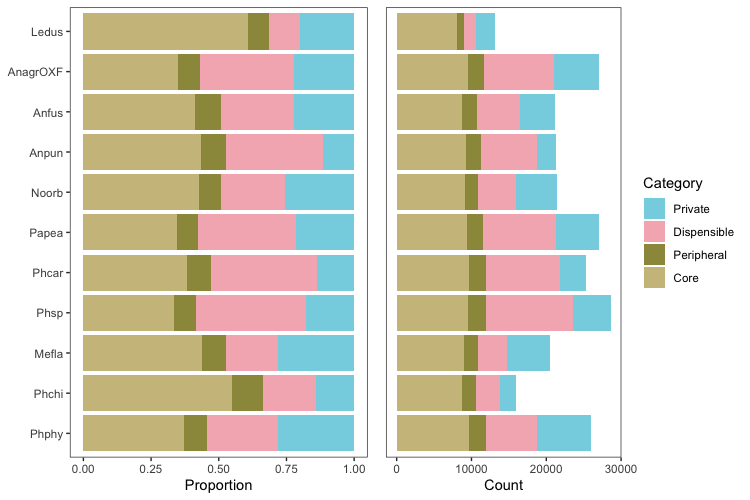
To make the bar charts of gene counts with in each category in, read in the categorized files, including the Orthogroups_UnassignedGenes.tsv file produced by OrthoFinder:
coreDF <- read.delim("Orthogroups.Core.tsv", header = T, sep ="\t")
periDF <- read.delim("Orthogroups.Peripheral.tsv", header = T, sep ="\t")
dispDF <- read.delim("Orthogroups.Dispensable.tsv", header = T, sep ="\t")
privDF <- read.delim("Orthogroups.Private.tsv", header = T, sep ="\t")
unassigned <- read.delim("Orthogroups_UnassignedGenes.txt", header = T, sep="\t")
# rename columns to more manageable names
colnames(coreDF) <- c("Orthogroups", "AnagrOXF", "Anfus", "Anpun", "Ledus", "Mefla", "Noorb", "Papea", "Phcar", "Phsp", "Phchi", "Phphy")
colnames(periDF) <- c("Orthogroups", "AnagrOXF", "Anfus", "Anpun", "Ledus", "Mefla", "Noorb", "Papea", "Phcar", "Phsp", "Phchi", "Phphy")
colnames(dispDF) <- c("Orthogroups", "AnagrOXF", "Anfus", "Anpun", "Ledus", "Mefla", "Noorb", "Papea", "Phcar", "Phsp", "Phchi", "Phphy")
colnames(privDF) <- c("Orthogroups", "AnagrOXF", "Anfus", "Anpun", "Ledus", "Mefla", "Noorb", "Papea", "Phcar", "Phsp", "Phchi", "Phphy")
colnames(unassigned) <- c("Orthogroups","AnagrBONN", "Anang" ,"AnagrOXF", "Anfus", "Anpun", "Ledus", "Mefla", "Noorb", "Papea", "Phcar", "Phsp", "Phchi", "Phphy")
Create the framework for a new dataframe summarizing all the gene categories.
Ours is an odd case because there will be some private genes created by removing A. agrestis Bonn and A. angustus from the analysis, so those are combined with the genes in Orthogroups_UnassignedGenes.tsv created by OrthoFinder.
species <- c(rep("AnagrOXF",4), rep("Anfus", 4), rep("Anpun", 4), rep("Ledus", 4), rep("Mefla", 4), rep("Noorb", 4), rep("Papea", 4), rep("Phcar", 4), rep("Phsp", 4), rep("Phchi", 4), rep("Phphy", 4))
geneType <- c(rep(c("Core", "Peripheral", "Dispensible", "Private"), 11))
abs <- as.numeric(c(sum(coreDF$AnagrOXF), sum(periDF$AnagrOXF), sum(dispDF$AnagrOXF), sum(privDF$AnagrOXF)+sum(nzchar(unassigned$AnagrOXF)),
sum(coreDF$Anfus), sum(periDF$Anfus), sum(dispDF$Anfus), sum(privDF$Anfus)+sum(nzchar(unassigned$Anfus)),
sum(coreDF$Anpun), sum(periDF$Anpun), sum(dispDF$Anpun), sum(privDF$Anpun)+sum(nzchar(unassigned$Anpun)),
sum(coreDF$Ledus), sum(periDF$Ledus), sum(dispDF$Ledus), sum(privDF$Ledus)+sum(nzchar(unassigned$Ledus)),
sum(coreDF$Mefla), sum(periDF$Mefla), sum(dispDF$Mefla), sum(privDF$Mefla)+sum(nzchar(unassigned$Mefla)),
sum(coreDF$Noorb), sum(periDF$Noorb), sum(dispDF$Noorb), sum(privDF$Noorb)+sum(nzchar(unassigned$Noorb)),
sum(coreDF$Papea), sum(periDF$Papea), sum(dispDF$Papea), sum(privDF$Papea)+sum(nzchar(unassigned$Papea)),
sum(coreDF$Phcar), sum(periDF$Phcar), sum(dispDF$Phcar), sum(privDF$Phcar)+sum(nzchar(unassigned$Phcar)),
sum(coreDF$Phsp), sum(periDF$Phsp), sum(dispDF$Phsp), sum(privDF$Phsp)+sum(nzchar(unassigned$Phsp)),
sum(coreDF$Phchi), sum(periDF$Phchi), sum(dispDF$Phchi), sum(privDF$Phchi)+sum(nzchar(unassigned$Phchi)),
sum(coreDF$Phphy), sum(periDF$Phphy), sum(dispDF$Phphy), sum(privDF$Phphy)+sum(nzchar(unassigned$Phphy))))
Combine elements into a dataframe. Set species and category factors to plot them in a certain order.
geneCountsDF<- data.frame(species,geneType,abs)
colnames(geneCountsDF) <- c("Species","Category","Count")
geneCountsDF$Species <- factor(geneCountsDF$Species, levels = c("Phphy","Phchi","Mefla","Phsp", "Phcar", "Papea", "Noorb", "Anpun", "Anfus", "AnagrOXF", "Ledus"))
geneCountsDF$Category <- factor(geneCountsDF$Category, levels = c("Private", "Dispensible", "Peripheral", "Core"))
Plot as stacked bar charts in two ways: absolute counts and as proportion of total.
percPlot <- ggplot(data = geneCountsDF, aes(x = Species, y = Count, fill = Category)) +
geom_bar(position='fill', stat="identity", show.legend = F) +
coord_flip() +
theme_bw() +
labs(x = "Proportion") +
scale_fill_manual(values = wes_palette("Moonrise3", type = 'discrete')) +
theme(panel.grid.major = element_blank(), panel.grid.minor = element_blank(), axis.title.y = element_blank())
absPlot <- ggplot(data = geneCountsDF, aes(x = Species, y = Count, fill = Category)) +
geom_bar(position='stack', stat="identity", show.legend = T) +
coord_flip() +
theme_bw() +
scale_fill_manual(values = wes_palette("Moonrise3", type = 'discrete')) +
theme(panel.grid.major = element_blank(), panel.grid.minor = element_blank(),axis.title.y=element_blank(), axis.text.y=element_blank(),axis.ticks.y=element_blank())
ggarrange(percPlot, absPlot, nrow=1)